- Indico style
- Indico style - inline minutes
- Indico style - numbered
- Indico style - numbered + minutes
- Indico Weeks View
Choose Timezone
Building on the success of previous editions, we are pleased to announce the upcoming FAIR-DI European Conference on Data Intelligence 2024. Following the remarkable achievements and insightful discussions at the 2020 and 2022 Conferences on a FAIR Data Infrastructure for Materials Genomics, we are excited to continue the discussion and further collaboration and innovation in this growing field with the theme of Data Intelligence.
Conference topics:
FAIR-DI Award for outstanding data handling in a PhD thesis:
 The FAIR-DI Award for data handling in a PhD thesis recognizes an excellent PhD thesis in the field of physics, materials science, or chemistry, which stands out by its exceptional handling of research data following FAIR principles. The recipient of this award will have the opportunity to present their thesis at the FAIR-DI European Conference on Data Intelligence 2024 and will receive a prize of €2000. Learn more about the award and apply here.
The FAIR-DI Award for data handling in a PhD thesis recognizes an excellent PhD thesis in the field of physics, materials science, or chemistry, which stands out by its exceptional handling of research data following FAIR principles. The recipient of this award will have the opportunity to present their thesis at the FAIR-DI European Conference on Data Intelligence 2024 and will receive a prize of €2000. Learn more about the award and apply here.
The arrival of materials science data infrastructures in the past decade has ushered in the era of data-driven materials science based on artificial intelligence (AI) algorithms, which has facilitated breakthroughs in materials optimisation and design. Of particular interest are active learning algorithms, where datasets are collected on-the-fly in the search for optimal solutions. We encoded such a probabilistic algorithm into the Bayesian Optimization Structure Search (BOSS) Python tool for materials optimisation [1]. BOSS builds N-dimensional surrogate models for materials’ energy or property landscapes to infer global optima, allowing us to conduct targeted materials engineering. The models are iteratively refined by sequentially sampling materials data with high information content. This creates compact and informative datasets. We utilised this approach for computational density functional theory studies of molecular surface adsorbates [2], thin film growth [3], solid-solid interfaces [4] and molecular conformers [5]. With experimental colleagues, we applied BOSS to accelerate the development of novel materials with targeted properties, and to optimise materials processing [7]. With recent multi-objective and multi-fidelity implementations for active learning, BOSS can make use of different information sources to help us discover optimal solutions faster in both academic and industrial settings.
[1] npj Comput. Mater., 5, 35 (2019)
[2] Beilstein J. Nanotechnol. 11, 1577-1589 (2020), Adv. Func. Mater., 31, 2010853 (2021)
[3] Adv. Sci. 7, 2000992 (2020)
[4] ACS Appl. Mater. Interfaces 14 (10), 12758-12765 (2022)
[5] J. Chem. Theory Comput. 17, 1955 (2020)
[6] MRS Bulletin 47, 29-37 (2022)
[7] ACS Sustainable Chem. Eng. 10, 9469 (2022)
FAIRmat provides research data management concepts and solutions for the field of solid-state physics. Its NOMAD portal has developed mature concepts and technological solutions for storing data according to the FAIR principles for selected theoretical and experimental data. Generalizing this approach is challenging due to the field’s diversity and complexity and due to missing standards.
In this presentation we present our comprehensive approach to establish FAIR data in the field of experimental solid-state physics despite its heterogeneity. The concept includes elaboration of standards, community building and methods that facilitate the community’s transition to FAIR standards. This includes our ongoing effort to establish a coherent ontology-based description, the FAIRmat-NeXus proposal, which is shared by the scientific community, but also by the instrument manufacturers.
The research data cycle typically starts with the planning of the experiment. At this stage, NOMAD CAMELS provides a low-threshold solution for configuring instrument control software instead of coding. Not only it simplifies the experimental protocol, it provides metadata-rich research data output along community-defined standards. Once the data are collected in an experiment, they can be straightforwardly fed into NOMAD Oasis, a local copy of NOMAD that can be tailored to serve the specific needs of individual labs. On this platform, one can work with the data, provide analyses and use it as local repository, with the option of transferring it to the world-wide NOMAD repository, where interoperability and finally a reuse of the data is prepared.
Hence, we establish solutions for the entire research data cycle. However, the complexity of solid-state physics imposes challenges. That is why we are actively promoting the establishment of data literacy in physics curricula with successful examples.
Efforts in materials science face challenges due to the heterogeneity and complexity of data sources, disparate data formats, and the need for standardized metadata. The NFDI-MatWerk ontology (MWO) [1] and the Materials Science and Engineering Knowledge Graph (MSE-KG) [2] aim to address these challenges by providing a unified framework for representing and integrating diverse data types and resources within the field. The MWO ontology has been developed as an extension of the NFDICore ontology [3, 4]. The NFDIcore ontology serves as a mid-level ontology, promoting interoperability among NFDI consortia by representing metadata about resources. It achieves this through mappings to standards like the Basic Formal Ontology (BFO), enhancing data harmonization and accessibility across the NFDI landscape. These ontologies serve the goal of facilitating seamless data interoperability, knowledge discovery, and collaboration across diverse materials science research domains. The MSE-KG v1.0 captures information on researchers, projects, institutions, software, workflows, instruments, publications, and datasets. In transitioning to the MSE-KG v2.0, integration with the research data graph presents challenges such as data interoperability, ontology alignment, and semantic integration.
Collaboration with domain experts is vital in overcoming these challenges, ensuring the seamless integration of domain-specific knowledge into the knowledge graph. The establishment of the NFDI-MatWerk linked open data (LOD) Working Group facilitates this collaboration, providing a platform for experts to contribute, share experiences, and collectively tackle the intricacies of LOD implementation in materials science. This collaborative effort is essential in shaping the future of data representation and accessibility in the MSE domain, ultimately maximizing the impact of scientific results and advancing research sustainability.
[1] http://purls.helmholtz-metadaten.de/mwo
[2] https://demo.fiz-karlsruhe.de/matwerk/
[3] https://doi.org/10.4126/FRL01-006474028
[4] https://github.com/ISE-FIZKarlsruhe/nfdicore
NFFA-DI (Nanoscience Foundries and Fine Analysis – Digital Infrastructure) is the NFFA upgrade for realizing a Full-Spectrum Research Infrastructure for nanoscience and
nanotechnology, capable of enhancing the Italian research competitiveness on the fundamental interactions of multi-atomic matter to
explore the origins of materials behaviour. The rationale of NFFA-DI is to integrate nanofoundry laboratories, i.e. facilities for atomically
controlled growth, structural characterization of nano-objects and nano-structured materials and the experimental facilities for the fine analysis of
matter using synchrotron radiation.
Inside the project, one challenging goal is realizing an integrated data management platform to
provide an integrated and unique set of services for all the user.
I will present the Overarching Fair Ecosystem for Data (OFED) at NFFA-DI,a flexible and modular modern architecture, how we have planned and how we are
developing and deploying it.It represents one of the paramount milestone in a scenario of new digital innovative IT approach, that integrates different
open source tools and collect, give access and share data accordingly to FAIR principles.
Data-driven materials discovery is coming of age, given the rise of 'big data' and machine-learning (ML) methods. However, the most sophisticated ML methods need a lot of data to train them. Such data may be custom materials databases that comprise chemical names and their cognate properties for a given functional application; or data may comprise a large corpus of text to train a language model. This talk showcases our home-grown open-source software tools that have been developed to auto-generate custom materials databases for a given application. The presentation will also demonstrate how domain-specific language models can now be used as interactive engines for data-driven materials science; The talk concludes with a forecast of how this 'paradigm shift' away from the use of static databases will likely evolve next-generation materials science.
In order to fulfill the interoperability requirement for FAIR research data, (meta)data need to comply with a community-agreed-upon language.
In the NOMAD Archive, materials science data are collected from heterogeneous sources, spanning synthesis, experimental characterization, and computations for modelling and analysis. This diversity necessitates flexible storage options, allowing users to create customized metadata schemas.
We present a strategic plan and a first version of a taxonomy for materials properties. A taxonomy is a terminology with structure, i.e., concepts are organized hierarchically in classes and subclasses. Crucially, it is expressed following the OWL2 (Web Ontology Language, version 2) which helps establish a standardized framework. This framework ensures that data from different sources are interoperable, meaning they can be combined, compared, and processed consistently across different systems and platforms.
The NOMAD taxonomy includes for each term: i) a curated, human-understandable definition, ii) a controlled list of synonyms, iii) the specification of the physical dimensions, and iv) the expected shape (tensorial rank). This taxonomy not only shapes the graphical interface of the NOMAD browser but also offers a controlled pool of predefined concepts. These can be reused in custom schemas, providing semantic guidance for newly defined metadata. By adhering to OWL2 standards and exportable in formats like RDF and TTL, our taxonomy facilitates seamless integration with linked data systems, enhancing global data exchange and collaboration within the materials-science community.
Finally, the taxonomy, so far limited to physical properties (i.e., the measured quantities), will be expanded to the description of the materials whose properties are measured and the characterization of experimental or computational methods that are used for performing the measurements.
NOMAD [nomad-lab.eu] [1] is an open-source, community-driven data infrastructure, focusing on materials science data. Originally built as a repository for data from DFT calculations, the NOMAD software can automatically extract data from the output of over 60 simulation codes. Over the past 2 years, NOMAD’s functionalities have been extensively expanded to support advanced many-body calculations, including GW, the Bethe-Salpeter equation (BSE), and dynamical mean-field theory (DMFT), as well as classical molecular dynamics simulations. Both standardized and custom complex simulation workflows not only streamline data provenance and analysis but also facilitate the curation of AI-ready datasets. In this contribution, we will show how these features, along with NOMAD’s adherence to the FAIR principles (Findability, Accessibility, Interoperability, Reusability) [2], provide a powerful framework for enhancing data utility and discovery [3]. In particular, the distinguishing characteristics of NOMAD from other Big-Data infrastructures will be highlighted through this FAIR-compliant perspective, e.g., the ability of users to clearly specify their own data quality needs. Finally, we will present an outlook, demonstrating NOMAD’s potential for creating a cohesive, interconnected scientific data landscape, where datasets can synergistically find a second life beyond their initial publications.
[1] Scheidgen, M. et al., JOSS 8, 5388 (2023).
[2] Wilkinson, M. D. et al., Sci. Data 3, 160018 (2016).
[3] Scheffler, M. et al., Nature 604, 635-642 (2022).
When gathering your research data and creating a knowledge graph, two aspects are key for achieving high data quality: making your data globally understandable and meaningful by semantic enrichment, and ensuring local conformance and completeness of your data by running validations. There are widely used languages within the Resource Description Framework (RDF) ecosystem to support these tasks, for example OWL to describe ontologies and SHACL to specify validations. However, applying these in your everyday work can be a very tedious and error prone task. Herbie, the hybrid electronic laboratory notebook and research database developed at Hereon, makes this process fast, simple and flexible.
Herbie is a client-server based web application wrapping an RDF triplestore and adding additional functionality on top of it. Its core design principle is to make use of these well-established frameworks in the RDF ecosystem like RDFS, OWL, SHACL, Schema.org, RO-Crate, etc., and give them an accessible and meaningful interface. In particular, we will see how Herbie builds on the SHACL Shapes Constraint Language to let you construct easily (re-)usable web forms helping you to turn your lab journal into a semantically rich knowledge base.
Additional features of Herbie include versioning of RDF graphs as well as managing access restrictions. Users can interact with Herbie using the graphical web interface or via its REST API to create, view, update or delete RDF graphs. This makes Herbie a platform for collaborative editing of a shared knowledge graph, applicable in various (research) environments.
An extensible open-source platform to support digitalization in materials science is proposed. The platform provides a modular framework for flexible web-based implementation of research data management strategies at scales ranging from a single laboratory to international collaborative projects involving multiple organizations.
The platform natively supports object types related to materials science, such as chemical systems and compounds. The extensible types system allows easy introduction of new user-defined object types. To implement deep integration with the types added, an external API is implemented, which is responsible for validation of documents, data extraction from them (for input in the database), and visualization of documents. Like late binding in programming, this allows the system to be extended without changing its source code by delegating the above-mentioned tasks to external web services configured at the object type level.
The system supports configurable templates for table data and properties for user-defined object types, allowing efficient storage and flexible search for material science entities. Build-in reports provide core metrics for data quantification and users/projects contribution evaluation. A special reporting API provides full read-only access to the system with the ability to securely execute arbitrary SQL queries to implement any form of custom reports or arbitrary data extraction for external use.
Multi-tenant support is implemented, enabling rapid deployment of new instances of the system, allowing the system to be provided as a Software-as-a-Service.
The system is here demonstrated as a data repository for combinatorial synthesis and high-throughput characterization data in Materials Discovery and Interfaces group, allowing efficient handling of thin film materials libraries and integration of data from different measurement devices with further flexible search capabilities. Furthermore, its use in the CRC TRR 247 is discussed.
This research was financially supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project-ID 388390466-TRR 247 (subproject INF).
The transition from specialist to generalist models in machine learning and deep learning represents a significant paradigm shift in addressing complex problems across various domains. Traditionally, specialist models have been developed for specific tasks within a particular field, relying heavily on domain knowledge and highly controlled datasets to optimize performance. While effective in their specialized areas, these models often lack the flexibility and scalability required for broader applications. In contrast, the advent of foundation models, such as transformers, has enabled the development of generalist models capable of tackling a diverse range of tasks without extensive task-specific customization.
In this work, we explore the role of physics and domain knowledge in this evolution, evaluating their importance in scientific problems such as defect analysis for electron microscopy and predicting structure-property relations. We discuss initial steps toward developing a foundation model to accelerate solar energy materials development, highlighting the peculiarities and limitations of real-world materials science data. This approach demonstrates that such models can be both data-efficient and capable of extrapolating beyond the training dataset - a crucial feature for scientific applications where data is relatively scarce or expensive to obtain.
Developing new materials requires extensive experimentation in synthesis and characterization, generating vast data sets. To keep this wealth of knowledge and adhere to the FAIR principles, effective data management is essential, involving standardized metadata schemas and integrated analysis tools. NOMAD has recently incorporated structured metadata schemas to manage experimental data from myriad sources, providing a user-friendly way of digitalizing experimental research.
In this contribution, we present a NOMAD analysis plugin, which establishes an integrated analysis workflow along with tools for automation. The plugin allows users to select and link structured data available in NOMAD from their experiments to perform analysis steps. Based on the data, specific standardized analyses may run automatically. The data entry associated with the analysis saves the settings along the output, making the analysis FAIR and thus promoting reproducibility. Additionally, it offers an open Python coding environment via Jupyter Notebooks for custom analysis adjustments.
The NOMAD analysis plugin leverages the platform's sharability and scalability while significantly enhancing its utility by enabling the training and integration of ML models. We illustrate its ML application with a case study on materials synthesis via metalorganic vapor-phase epitaxy. By enabling automated analysis, ensuring reproducibility, and supporting ML applications, the plugin motivates researchers to digitalize their workflows, effectively reducing adoption barriers.
Research data management i(RDM) has been receiving much attention, being in the focus of many institutes often upon pressure from funding agencies and by thriving for good scientific practice.
Several solutions are being developed, mostly focusing on central database systems allowing for structured data storage. These solutions allow for classification, access control, publishing of data. Some (such as the NOMAD system) also provide in-system data analysis.
However, a major part of the data and information originate from local personal computers. Depending on resources, such as computer facility, network bandwidth, etc., this local storage plays an often ignored, but organic part in the RDM. And it is this part where perhaps most information gets lost due to difficulties in keeping consistent storage practices.
(Another important aspect is that utilizing local storage, small institutes with limited resources can still maintain a several terrabytes large distributed data store.)
Here I propose the usage of a simple software that helps the user seeing a simplified folder tree for project management, as well as easy listing of experiment / data related metadata information. The RDM-desktop project aims to be a simple and easy tool only for this purpose not limiting the user in her/his routine activities. It is aimed to be simple and light-weight following the KISS development principles.
Material databases contain vast amounts of information, often harboring intricate connections and dependencies within material systems, some of which may remain undiscovered. Their structured organization naturally lends itself to the application of machine learning techniques. Through machine learning, we can unlock the tools necessary to discover potentially hidden structure-property relationships.
In this study, we present a use case where generic interactive workflows employ machine learning to automatically uncover such linkages from the research data infrastructure Kadi4Mat (https://kadi.iam.kit.edu/).
Within this infrastructure, data from simulated and experimental analyses of material systems are stored using a unified metadata scheme for coherent structuring.
In our use case, we first extract this uniformly structured data from the Kadi4Mat platform and prepare it for use in machine learning methods.
We then iteratively train neural networks to identify correlations between the microstructural compositions of the investigated materials and their resulting macroscopic properties. Using explainable AI techniques, in particular layer-wise relevance propagation, we identify the most influential parameters governing macroscopic properties. This allows us to streamline our neural network to focus only on key microstructural features to predict macroscopic properties.
Ultimately, we refine our network to predict macroscopic properties from minimal inputs, yielding a comprehensive material property map. This map concisely summarises the results of our network, allowing the macroscopic properties of a material to be quickly and easily determined from its microstructural composition.
This streamlined approach speeds up the materials research process and facilitates a data-driven accelerated development of new materials by providing researchers with invaluable insights into structure-property relationships.
In materials development, creating new data points is often very costly due to the effort needed for materials synthesis, sample preparation and characterization. Therefore, all available knowledge in terms of data, physical models and expert knowledge should be exploited in the most efficient way (optimal knowledge exploitation). Moreover, the number of new samples/data points to be produced in terms of synthesis and characterization of new materials should be kept at a minimum to save time and money (sample efficiency). The ALPmat is an Active Learning Platform for MATerials design, targeted at optimal knowledge exploitation and sample efficiency. For optimal knowledge exploitation, a hybrid approach is followed, where physical models and expert knowledge are combined with data from observations. The resulting hybrid models are used for an Active Learning Loop (ALL) to improve the addressed properties in an iterative way via optimization of the material’s chemistry and processing conditions, while also minimizing the number of new samples ensuring sample efficiency. We will present the details of the ALPmat in terms of hard- and software for the platform backbone, the FAIR database, the framework for running physical modeling and Bayesian optimization algorithms, and integrated software services. Moreover, we will present data models for our use cases. These data models are linked to the state of the investigated sample and are used to uniquely identify use case-specific synthesis, processing and characterization steps. They serve as a basis for the ingestion of comprehensive metadata and ensure that the data are fully FAIR. Finally, we will show the first results for the application of the developed methodology and infrastructure for the use case of bainitic steels. For this use case, we are performing a multi-objective optimization of the uniform elongation and the yield strength as a function of chemical composition and processing conditions.
The field of chemical sciences has seen significant advancements with the use of data-driven techniques, particularly with large datasets structured in tabular form.
However, collecting data in this format is often challenging in practical chemistry, and text-based records are more commonly used [1]. Using text data in traditional machine-learning approaches is also difficult. Recent developments in applying large language models (LLMs) to chemistry have shown promise in overcoming this challenge. LLMs can convert unstructured text data into structured form and can even directly solve predictive tasks in chemistry. [2, 3] In my talk, I will present the impressive results of using LLMs, showcasing how they can autonomously utilize tools and leverage structured data and “fuzzy” inductive biases. To enable the training of a chemical-specific large language model, we have curated a new dataset along with a comprehensive toolset to utilize datasets from knowledge graphs, preprints, and unlabeled molecules. To evaluate frontier models trained on such a dataset, we specifically designed a benchmark to evaluate the chemical knowledge and reasoning abilities. I will present the latest results, demonstrating the potential of LLMs in advancing chemical research. [4]
References:
[1] Jablonka, K. M.; Patiny, L.; Smit, B. Nat. Chem. 2022, 14 (4), 365–376.
[2] Jablonka, K. M; et al. Digital Discovery 2023, 2 (5), 1233–1250.
[3] Jablonka, K. M.; Schwaller, P.; Ortega-Guerrero, A.; Smit, B. Leveraging large language models for predictive chemistry. Nat. Mach. Int. 2024, 6, 161–169.
[4] Mirza, A.; Alampara, N.; Kunchapu, S.; Emoekabu, B.; Krishnan, A.; Wilhelmi, M.; Okereke, M.; Eberhardt, J.; Elahi, A. M.; Greiner, M.; Holick, C. T.; Gupta, T.; Asgari, M.; Glaubitz, C.; Klepsch, L. C.; Köster, Y.; Meyer, J.; Miret, S.; Hoffmann, T.; Kreth, F. A.; Ringleb, M.; Roesner, N.; Schubert, U. S.; Stafast, L. M.; Wonanke, D.; Pieler, M.; Schwaller, P.; Jablonka, K. M. Are Large Language Models Superhuman Chemists? arXiv 2024. https://doi.org/10.48550/ARXIV.2404.01475.
X-ray absorption spectroscopy (XAS) is one of the characterisation techniques which can be employed to probe electronic structure as well as local structure of functional materials. XAS data analysis involves comparison with theoretical or experimental references and processing of the data includes steps, i.e., calibration, background subtraction, normalization etc. Thus, for the extraction of usable information from any XAS measurement, users need to have access to both high quality reference spectra with documented metadata fields and standard analysis tools. Considering these requirements, we have established XAS reference database under DAPHNE4NFDI called RefXAS, where users are provided with well curated XAS reference spectra along with related metadata fields and online processing tools for visualizing the data at the interface. For the present database, we have categorized meta data fields under “Sample”, “Spectra”, “Instrument” and “Bibliography”, and further sub-fields were defined under these categories. As an important aspect of a curated database users would be able to easily judge the quality and the usability of each data set by looking at the quality criteria formulated for any reference data which has been uploaded to the database. Further, standardization of data formats has been one of the challenges in the XAS community. In this regard, the interface of RefXAS database has been tested with different data/file formats so that users would be able to upload the data in different formats as received from experimental facilities which includes synchrotron beamlines as well as laboratory instruments. In the present work, the significance of metadata fields for the reusability as well as reproducibility of results (FAIR data principle) has been discussed. The formulation of quality criteria for the data uploaded at the database have been examined and usability/interoperability of available XAS data/file formats have been explored.
Abstract:
With advancements in the sensitivity of present synchrotron facilities and the refinement of analytical methods, X-ray based techniques have become a standard approach for the structural characterization of intricate solid material systems. X-ray absorption spectroscopy (XAS) stands out as one of the most effective methodologies utilized for the analysis of various functional materials. Within DAPHNE4NFDI, RefXAS serves as a pioneering reference database in the field of XAS, developed to enhance scientific research through a sophisticated platform for both submitting and accessing high-quality XAS data. Analysis of XAS data requires comparison with previously measured experimental/simulated reference spectra. Reference data is essential for data evaluation and therefore, a curated database that offers high-quality reference data is required. In order to achieve that, our system has the ability to execute quality control in the background, i.e. during uploading, predefined quality criteria are checked automatically. Furthermore, RefXAS includes raw and processed data, an intuitive interface for uploading and evaluating the data along with their comprehensive metadata via a dedicated website. Our database supports various data formats from diverse XAS setups, including synchrotron and laboratory instruments, which aids in standardizing data handling within the community while promoting FAIR principles. The classification of the metadata fields for reporting/uploading any data enhances the traceability and usability of data. The current prototype features a human verification procedure, currently being tested with a new user interface designed specifically for curators, a user-friendly landing page, a full listing of datasets, advanced search capabilities, a streamlined upload process, and finally, a server-side automatic authentication and (meta-)data storage via MongoDB, PostgreSQL, and and (data-) files via relevant APIs. In the present work, the different features of the RefXAS interface have been presented.
Automating instrumentation is a big challenge for any lab. In established labs, there is often large amount of existing infrastructure, with the benefits of automation only tangible after several components of a set-up are automated. In smaller labs, automation is often hampered by lack of personnel and know-how.
Here, we present tomato, an open-source, python-based, cross-platform instrument automation framework that is part of the dgbowl suite of tools for digital (electro)-catalysis. With tomato, an incremental approach to instrument automation is possible, as tomato outputs data in NetCDF format. This means tomato's outputs are fully consistent with the standardised data output format of our FAIR data parser, yadg.
We present two case studies for tomato: our work on the integration of off-the-shelf battery testing infrastructure with a workflow management toolkit as part of the Aurora project, and our progress in incremental automation of our cavity operando conductivity set-up, COCoS. The case studies illustrate how painless it is to both deploy tomato as well as integrate it in more complex workflows.
The advent of data-driven approaches in materials science requires the aggregation of heterogeneous data from various sources, including simulation and experiments, which span different length scales and encompass a wide range of compositions, structures and thermodynamic conditions. In materials design, a major challenge arises from the combination of different software and file formats, leading to interoperability issues. To achieve workflow and data reusability, and meaningful interpretation, it is crucial to ensure well-described (meta)data at each step of the simulation workflow. Our aim is to establish a machine-readable standard for representing material structures, workflows and calculated properties, including their intrinsic relationships.
To describe simulations at the atomistic level, we have developed an ontology for computational material samples, CMSO, complemented by ontologies for crystallographic defects, which are often neglected in standardization approaches. Another essential aspect to achieve interoperability is describing the simulation method, this is facilitated by the Atomistic Simulation Methods Ontology (ASMO). Data annotation using these ontologies is embedded directly in the workflow with the software atomRDF. This allows users to semantically annotate jobs using pyiron as an example for the workflow environment and build an application-level knowledge graph.
We demonstrate the benefits of such a knowledge graph for: (i) aggregating data from heterogeneous sources in a scale-bridging fashion, (ii) allowing complex queries through an automated system to explore the data; (iii) identifying new trends and extracting material properties that were not explicitly calculated. We illustrate these benefits with two examples: the calculation of formation energies of crystal defects and the extraction of thermodynamic quantities from existing simulation data. This innovative approach, combining simulation workflows and semantic technologies, accelerates the analysis, sharing and reuse of data. Leveraging the advantages of a knowledge graph enhances interoperability and data quality, increasing compliance with the FAIR principles.
Photoemission spectroscopy (PES) is presented as a use case for pioneering future research data concepts. We will show how FAIR research data can be organized and how we intend to create benefits for the participating scientists. We will present an extensive and elaborated standard (NXmpes) for harmonizing PES data using NeXus. This standard is developed in collaboration with the PES community and hardware companies in the field to facilitate the integration of FAIR data into research labs.
To demonstrate the potential of our approach, we present a workflow and data pipeline derived from time- and angle-resolved photoemission spectroscopy. We show how such a pipeline can be integrated into NOMAD, a research data management and publication software developed in FAIRmat. As an alternative approach, we demonstrate stand-alone tools for generating FAIR data, facilitating their integration into custom data generation pipelines. We also present our strategy of working with leading PES instrument manufacturers to promote interoperability of NXmpes with their software solutions, and how this approach benefits scientists in their labs.
Low temperature Scanning Tunneling Microscopy (STM) and Spectroscopy (STS) provide important insights to materials properties which should be captured and stored following the FAIR principles. We have developed a data format proposal for the NeXus community standard which provides a rich vocabulary for representing all important experimental data and metadata. An end-to-end solution embedded into the NOMAD[1] research data management platform is presented which also demonstrates the research experience as the new standard is in a daily use in real-life laboratory environment.
With the help of NeXus[2] that provides a flexible data modelling platform with a community standardisation process, we created the generic data model NXsts[3] which supports both STM and STS experiments as well as the special needs for covering experiments performed at low temperature. Additionally to modelling experimental data and related metadata, the NXsts vocabulary also supports handling data-analysis results (e.g. topography and dI/dV). In this work, we show how experimental data can be converted into the NeXus standard and used efficiently in NOMAD.
References
[1] Scheidgen et al., (2023). NOMAD: A distributed web-based platform for managing materials science research data. Journal of Open Source Software, 8(90), 5388, https://doi.org/10.21105/joss.05388
[2] Könnecke, M., et al., The NeXus data format, J. Appl. Crystallogr. 2015, 48, 1, 301-305.
[3] https://fairmat-nfdi.github.io/nexus_definitions/classes/contributed_definitions/NXsts.html#nxsts
Keywords
FAIR data, NOMAD, NeXus, scanning probe, STM, STS
In modern material science the amount of generated experimental data is rapidly increasing while analysis methods still require many manual work hours. Especially, this is the case for X-ray photoelectron spectroscopy (XPS), where quantification is a complex task and, in many cases, can be properly done by experts only. However, these problems could be overcome by the use of a neural network-based approach (NNA). An important question is to validate NNA-based results and to compare them with results obtained with a use of a commonly used, manual fitting procedure. Since the available experimental data is insufficient for network training, a synthetic dataset was created using parameters obtained from the real experimental XP spectra measured on a reference sample. A 4-component model has been chosen and some parameters (binding energies, FWHM) were almost fixed, but the peak intensity was a free parameter keeping the total area of all 4 components as constant (normalization). As commonly used fitting procedure CasaXPS software was applied. After training on the synthetic data, the neural network was tested on the experimental data obtained from another sample and predicted area percentages were compared with the fitting results. The predicted area percentages are in good agreement with corresponding area percentages from the fitting with CasaXPS. Moreover, it was established that it is crucial to choose a proper model and corresponding NNA training set, otherwise the experimental data could not be evaluated properly. It means that this approach can therefore be successfully used not only for XPS quantification tasks directly, but also to validate proposed models.
Achieving an interoperable representation of knowledge for experiments and computer simulations [1-4] is the key motivation behind the implementation of tools for FAIR research data management in the condensed-matter physics and materials engineering communities. Electron microscopy and atom probe tomography are two key materials characterization techniques used globally and across disciplines. Many research data artifacts from these communities are already publicly shared using various formats but offering limited interoperability [5, 6]. This highlights the need for the development of tools specialized in information extraction and semantic mapping. Fundamental to these tools' effectiveness is the creation of thorough and transparent documentation using outlets which are readily available to the public, and derived from collaborative efforts where community representatives concur on establishing and employing standardized forms of knowledge representation.
In this work, we report on our progress on developing comprehensive data schemas, respective domain ontologies, and software tools for generating interoperable research data artifacts within the electron microscopy and atom probe tomography communities. Technically, these tools are standalone software libraries, plugins, and data schemas that we have incorporated into NOMAD Oasis [5-7], offering a locally-installable version of the NOMAD research data management system (RDM). This integration aims at an augmentation of the RDM capabilities in note-keeping, file format parsing, cloud-based domain-specific data analyses, and information retrieval with greater customizability for specific research needs.
We will present specific examples of customizations for electron microscopy, atom probe tomography, microstructure evolution modeling [6-9], and how these can be used out-of-the-box in NOMAD.
1 M. D. Wilkinson et al., (2016), https://doi.org/10.1038/sdata.2016.18
2 A. Jacobsen et al., (2020), https://doi.org/10.1162/dint_r_00024
3 M. Barker et al., (2022), https://doi.org/10.1038/s41597-022-01710-x
4 M. Scheffler et al., (2022), https://doi.org/10.1038/s41586-022-04501-x
5 M. Scheidgen et al., (2023), https://doi.org/10.21105/joss.05388
6 https://github.com/FAIRmat-NFDI/
7 https://gitlab.mpcdf.mpg.de/nomad-lab/nomad-FAIR
8 https://www.re3data.org
9 https://explore.openaire.eu
Within the expansive domain of Metal-Organic Frameworks (MOFs), navigating the vast datasets for impactful research has posed significant challenges. Addressing this, our study introduces a groundbreaking methodology through MOFGalaxyNet, employing Social Network Analysis (SNA) to illuminate the structure and dynamics of MOF interactions. The core of our strategy, the Black Hole approach, identifies the most influential MOFs—akin to celestial black holes for their significant pull on surrounding entities. This leads to creating the Black Hole dataset, a curated collection of MOFs identified for their pivotal roles within the network. Through sophisticated SNA, we extract the Black Hole dataset, a concise yet comprehensive assembly of influential MOFs poised for significant breakthroughs in research. The Black Hole dataset, derived from advanced community detection and centrality analysis, also provides a focused, high-value resource for ML applications in MOF research. Utilizing the Girvan-Newman algorithm, we segment MOFGalaxyNet into communities, employing Degree and Betweenness centrality measures to highlight key MOFs. The resultant Black Hole dataset not only streamlines research focus towards MOFs with the highest potential impact but also embodies the FAIR (Findable, Accessible, Interoperable, Reusable) principles, offering a robust foundation for ML-driven advancements in materials science. Applying the Girvan-Newman algorithm for community detection, alongside Degree and Betweenness centrality analyses, facilitates identifying and categorizing Black Hole MOFs within MOFGalaxyNet. This methodology empowers ML in MOF Research by providing ML practitioners in the MOF community with a data-rich, targeted, and technically vetted resource for predictive modeling and algorithm training.
The FAIR principles (Findable, Accessible, Interoperable, Reusable) serve as a reference for assessing the quality of data storage and publication [1]. NOMAD [nomad-lab.eu] [2, 3] is an open-source data infrastructure for materials science data that is built upon these principles. In this contribution, we will demonstrate the interplay between high-quality data and knowledge using the functionalities provided by NOMAD and with DFT as an example case. In particular, we will showcase the dynamic and flexible metadata framework, designed for a clearer, more customizable navigation of the zoo of density functionals. We will then show how precision and accuracy metrics are represented within this framework, and how they can be linked to benchmark datasets. Finally, we will present a brief outlook on the future of NOMAD as a platform that fosters an interconnected research community and engaged scientific discourse.
[1] Wilkinson, M. D. et al., Sci. Data 3, 160018 (2016).
[2] Scheffler, M. et al., Nature 604, 635-642 (2022).
[3] Scheidgen, M. et al., JOSS 8, 5388 (2023).
Recent discoveries in astroparticle physics, including cosmic accelerators, gravitational waves from black-hole mergers, and astronomical neutrino sources, underscore the importance of a multi-messenger approach. The transient and rare nature of these astrophysical phenomena necessitates interdisciplinary work with diverse modern and historical data, emphasizing the need for FAIR (Findable, Accessible, Interoperable, and Reusable) data management.
Founded in 2013, the KASCADE Cosmic-ray Data Centre (KCDC) was a pioneer in publishing comprehensive data from the KASCADE-Grande experiment and adopting recent data curation trends. Today, KCDC is a web-based platform for high-energy astroparticle physics, offering open access to datasets from experiments like KASCADE-Grande, LOPES, Maket-Ani and others. These datasets are available in widely used formats, accessible via a web portal or an API, and enriched with both high-level and discipline-specific metadata to enhance findability and interoperability. Beyond serving as a data archive, KCDC provides a wide range of other digital resources, including cosmic ray energy spectra, simulations, tutorials, and Jupyter Notebooks, supported by a JupyterLab-based online analysis platform. Our current efforts focus on enriching these digital objects with machine-readable metadata and developing a unified metadata schema to standardize data management across the platform. This approach aims to simplify curation and align KCDC metadata with the standards of partner platforms, such as the PUNCH4NFDI Data Platform. Collaboration through initiatives like PUNCH4NFDI and NAPMIX on advanced metadata standards and tools supports interdisciplinary research, enabling KCDC to continually improve its data management strategies and broaden access to its digital resources.
This presentation will discuss data management on the KCDC platform, the metadata tools in use, and ongoing metadata schema developments.
Optical spectroscopy covers experimental techniques such as ellipsometry, Raman spectroscopy, or photoluminescence spectroscopy. In the upcoming transformation process of the research environment towards FAIR data structures, these techniques will play a crucial role as they govern various fundamental and easily accessible material properties such as reflectivity, light absorption, bandgap, or material composition. The present barriers to this transformation are indicated and a standard to surpass these problems is pointed out. This was done in cooperation with hardware companies as well as scientists. The advantage and potential of these resulting FAIR data structures are highlighted as well as their integration in research labs.
This is done exemplarily for the optical spectroscopy standard and one of its respective specializations. The processing steps are shown to transform the nowadays-usual measurement setups into FAIR data setups.
The emergence of big data in science underscores the need for FAIR (Findable, Accessible, Interoperable, Reusable) [1] data management. NOMAD [nomad-lab.eu] [2, 3] is an open-source data infrastructure that meets this demand in materials science, enabling cross-disciplinary data sharing and annotation for both computational and experimental users. In this contribution, we will present our recent work in extending NOMAD to support a range of many-body and excited state calculations, including GW, BSE, and DMFT, among others. We will demonstrate how NOMAD captures these workflows in an automated but flexible fashion, enabling findability and clear, visual overviews. Finally, we will present an outlook on NOMAD′s potential for large-scale interoperability and harmonization between computational and experimental data in the field of spectroscopy.
[1] Wilkinson, M. D. et al., Sci. Data 3, 160018 (2016).
[2] Scheffler, M. et al., Nature 604, 635-642 (2022).
[3] Scheidgen, M. et al., JOSS 8, 5388 (2023).
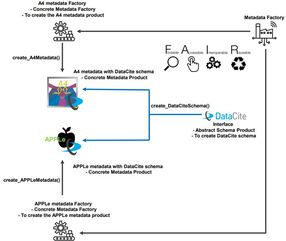
The PATOF project builds on work at MAMI particle physics experiment A4. A4 produced a stream of valuable data for many years which already released scientific output of high quality and still provides a solid basis for future publications. The A4 data set consists of 100 TB and 300 million files of different types (Vague context because of hierarchical folder structure and file format with minimal metadata provided 1.). In PATOF we would like to build a “FAIR Metadata Factory 2”, i.e. a process to create a naturally evolved metadata schema that can be used across research fields. The first focus will be on creating machine-readable XML files containing metadata from the logbook and other sources and to further enrich them 3.
In PATOF, we intend to conclude the work on A4 data, to extract the lessons learned there in the form of a cookbook that can capture the methodology for making individual experiment-specific metadata schemas FAIR, and to apply it to four other experiments: The ALPS II axion and dark matter search experiment at DESY. The PRIMA experiment at MAMI in Mainz for measuring the pion transition form factor. The upcoming nuclear physics experiment P2 at MESA in Mainz. Finally, the LUXE experiment at DESY planned to start in 2026. The focus of PATOF is on making these data fully publicly available.
The objectives of the project are i) a FAIR Metadata Factory (i.e. a cookbook of (meta)data management recommendations), and ii) the FAIRification of data from concrete experiments. Both aspects are inherently open in nature so that everybody can profit from PATOF results. The cookbook is expected to be further enhanced with contributions from other experiments even after PATOF (“living cookbook”).


There has been a distinct lack of FAIR data principles in the field of photoemission spectroscopy (PES). Within the FAIRmat consortium, we have been developing an end-to-end workflow for data management in PES experiments using NOMAD and NeXus, a community-driven data-modeling framework for experiments [1]. We will present an extensive and elaborated standard (NXmpes) for harmonizing PES data using NeXus. Specifically, we present our strategy to collaborate with leading manufacturers of PES equipment as well as the wider PES community to foster interoperability of NXmpes with existing measurement protocols and software solutions.
Finally, we provide an example for establishing a FAIR data pipeline using NXmpes in conjunction with the NOMAD research data management platform with a focus on X-ray photoelectron spectroscopy (XPS) data. We will show how research workflows and analysis results in XPS are represented in NOMAD and how individual scientists as well as the wider XPS community benefit from adapting these data pipelines.
References
[1] Könnecke, M., et al., The NeXus data format, J. Appl. Crystallogr. 2015, 48, 1, 301-305.
[2] https://fairmat-nfdi.github.io/nexus_definitions/classes/contributed_definitions/NXmpes.html
In order to achieve interoperability for data of different origin, FAIRmat is contributing to the materials science data management platform, NOMAD. It features flexible, but structured data modeling, allows custom data ingestion, while providing efficient search capabilities and online visualization of datasets. Several standard data formats are supported by NOMAD including the NeXus format to support experimental data from various techniques.
In this work, NeXus is presented as a standardisation platform for community-driven data modeling for experiments. We will report on recent progress where scientific community and technology partners have joined forces to achieve new standards, and also report on how these new standards are integrated in NOMAD.
Keywords
FAIR data, standardization, ontology, research data management
Molecular dynamics simulations combined with first-principles calculations have long been the gold-standard of atomistic modeling, but have also been associated with steep computational cost, and with limitations on the accessible time and length scales. Machine-learning models have greatly extended the range of systems that can be studied, promising an accuracy comparable with that of the first-principles reference they are fitted against.
Given the interpolative nature of machine-learning models, it is crucial to be able to determine how reliable are the predictions of simulations that rely on them, as well as to understand the physical underpinnings -- if any -- for the successes and failures of different frameworks.
I will discuss a few examples of how understanding the mathematical structure of ML models helps to use them to interpret the outcome of atomistic simulations, in terms of familiar concepts such as locality, range and body order of interactions.
Then, I will give a brief overview of the different approaches that are available to obtain a quantitative measure of the uncertainty in a machine-learning prediction, and discuss in particular an inexpensive and reliable scheme based on an ensemble of models. By a scheme that we refer to as "direct propagation of shallow ensembles" (DPOSE) we estimate not only the accuracy of individual predictions, but also that of the final properties resulting from molecular dynamics and sampling based on ML interatomic potentials.
State-of-the-art Bayesian optimization algorithms have the shortcoming of relying on a rather fixed experimental workflow. The possibility of making on-the-fly decisions about changes in the planned sequence of experiments is usually excluded and the models often do not take advantage of known structure in the problem or of information given by intermediate proxy measurements [1-3]. We hypothesize that an extended Bayesian optimization procedure, with surrogate models and acquisition functions that can flexibly choose to modify the workflow on the fly, will improve the performance of state-of-the-art methods for optimization in self-driving labs.
To address these limitations, we developed a surrogate model composed of a sequence of Gaussian processes, that can take advantage of the modular structure of experimental processes to handle sparse datasets where only partial information (proxy measurements) is available [4]. We implemented an acquisition function, based on a mixture of expectation improvement and upper confidence bound, that allows the optimizer to selectively sample from individual sub-processes. Finally, we devised a synthetic dataset generator to simulate multi-step processes with tunable function complexity at each step, to evaluate the efficiency of our model compared to standard BO under various scenarios.
We conducted experiments to evaluate our model across nine distinct scenarios. In all scenarios our multi-step optimizer outperformed the benchmark methods, demonstrating superior performance in terms of both the quality of the optimum and in terms of convergence speed. This advantage is particularly evident in scenarios where the complexity of the first step exceeds that of the second step. We are currently in the process of validating our results on real-world datasets.
[1] Wu et al. 2023, JACS 145 (30).
[2] Seifermann, et al. 2023. Small Methods, 7(9).
[3] Jenewein et al. 2023. Journal of Materials Chemistry A, 12(5).
[4] Torresi et al. 2024, submitted.
Nanophotonic structures that enhance light-matter interaction can increase the sensitivity of spectroscopic optical measurements, such as detection and enantiomer discrimination of chiral molecules. However, this improved sensitivity comes at the cost of complicated modification of the spectra, and it is necessary to account for this during the experiment and in data analysis. This calls for the construction of a digital twin: a comprehensive computer model of the experiment.
In this work [1], we develop a digital twin for a chiral sensing platform based on helicity-preserving optical cavities that enhance the circular dichroism (CD) signal of molecules [2]. The digital twin comprises a series of simulations, each related to a certain part of the sensing device. The chiral molecules are modelled using density functional theory-based simulations, and the light-matter interaction and the formation of the detectable signal are modelled using optical simulations [3,4]. A machine learning-based approach bridges the calculation results with experimentally measurable data. The digital twin is needed to interpret the experimental results and reconstruct the molecule’s CD spectrum from measurement data. It is also used to design the optical cavities while accounting for the limitations of the experimental equipment.
The idea of using a digital twin to support nanophotonically enhanced optical experiments broadly applies to measurements other than CD spectroscopy. As increasing effort is put into utilizing nanophotonic concepts in measurement devices, we expect digital twins to be an important part of such experimental workflows.
[1] M. Nyman et al., Laser Photonics Rev. 2024, 2300967 (2024).
[2] J. Feis et al., Phys. Rev. Lett. 124, 033201 (2020).
[3] I. Fernandez-Corbaton et al., ChemPhysChem 21, 878 (2020).
[4] D. Beutel et al., Comp. Phys. Comm. 297, 109076 (2024).
In the rapidly evolving field of materials science, the shift towards data-centric research needs enhanced strategies for data management, sharing, and publication. This presentation introduces NOMAD (https://nomad-lab.eu), a web-based platform developed by the NFDI consortium FAIRmat. Designed to address these challenges, NOMAD pioneers the application of FAIR principles (Findable, Accessible, Interoperable, and Reusable) to materials science data, thereby facilitating a more efficient, open and collaborative research environment in a federated infrastructure fashion. The core focus of this talk is the striking changes NOMAD has undergone in transitioning from an archive and repository for ab-initio calculations to a global platform for managing materials science data. I will introduce NOMAD Oasis, a locally installable and customizable version of this platform designed to enable the creation of FAIR data from its inception, while simultaneously becoming nodes in a rapidly expanding network of interconnected data hubs. These platforms support a broad spectrum of data-driven research activities within materials science. I will showcase how NOMAD's infrastructure serves as a critical backbone for data-driven research across various domains. These include the accelerated synthesis of materials via physical vapor deposition methods, complex computational workflows, big data strategies for developing novel solar cells, hosting databases for experimental heterogeneous catalysis and metal-organic frameworks, and powering the application of generative AI in materials research.
Data Science (DS) is a multidisciplinary field combining different aspects of mathematics, statistics, computer science, and domain-specific knowledge to extract meaningful insights from diverse data sources. DS and AI involve various artifacts, e.g., datasets, models, ontologies, code repositories, execution platforms, repositories, etc. The NFDI4DataScience (NFDI4DS) project endeavors to enhance the accessibility and interoperability of research data in the NFDI and DS domain. It achieves this by linking digital artifacts and ensuring their FAIR (Findable, Accessible, Interoperable, and Reusable) accessibility, thereby fostering collaboration across various DS and AI platforms. To this end, the NFDI4DS Ontology is built upon the common NFDI core ontology that is mapped to the Basic Formal Ontology to enable interoperability [1,2].
The NFDI4DS ontology is a mid-level ontology describing all resources (datasets, data providers, persons, projects, and other entities) within the data science domain of NFDI4DS. Moreover, the ontology forms the basis for two knowledge graphs: the Research Information Graph (RIG) and the Research Data Graph (RDG). RIG covers metadata about the NFDI4DS consortium’s resources, persons, and organizations whereas RDG covers content-related index data from the consortium’s heterogeneous data resources. RIG serves as a backend for the web portal that enables interactive access and management of this data. Both RIG and RDG will be made available and searchable using the NFDI4DS Registry platform. Furthermore, the NFDI4DS consortium also aims to collaborate with other NFDI consortia for further seamless integration of domain-specific knowledge into the RDG.
[1] Oleksandra Bruns, Tabea Tietz, Etienne Posthumus, Jörg Waitelonis, Harald Sack. NFDIcore Ontology. Revision: v2.0.0. Retrieved from: https://nfdi.fiz-karlsruhe.de/ontology/2.0.0
[2] Tietz, Tabea, et al. "From Floppy Disks to 5-Star LOD: FAIR Research Infrastructure for NFDI4Culture." 3rd Workshop on Metadata and Research (objects) Management for Linked Open Science (DaMaLOS), co-located with ESWC. 2023.
Self-assembling peptides (SAPs) are a type of biomaterial consisting of short aminoacid sequences that can be controlled under specific physicochemical conditions. SAPs form nanostructures that can mimic biological scaffolds giving them numerous applications such as in drug delivery, tissue engineering, biosensors, etc.
In this project we will create new SAP sequences based on desired biophysicochemical properties. Not only has deep learning proven adept at this task, it may also help us shed some light on the correlation between peptide sequence, structure and biological activity.
However the quality of the result heavily depends on the number and quality of the training data. It is therefore paramount to construct a database of known peptide sequences and their biophysicochemical properties obtained by our and other groups from wet lab experiments and complemented with computational methods. The database should be open and possess a user-friendly interface adapted to professionals in the fields of chemistry and biology that might not have experience in working with extensive datasets. It should deal with both entries from automated experimental setups, as well as manual input from a variety of users. The data structure should be consistent. Changes to specific data may only be carried by the user who originally uploaded the data and people whom the user specified as collaborators. These data changes must be traceable.
Many phenomena and functional devices in optics and photonics rely on discrete objects, called scatterers, that interact with light in a predefined way. The optical properties of these scatterers are entirely described by the T-matrix. The T-matrix is computed for a given scatterer from a larger number of solutions to the Maxwell equations. Still, once known, various photonic materials made from these scatterers can be semi-analytically studied within a multi-scattering formalism. These photonic materials can consist of periodically or many, i.e., up to millions, a-periodically arranged objects with known T-matrices. Such a usage scenario points to the importance of storing the T-matrices for future exploitation since the computation of the T-matrix is demanding. Recalculating a T-matrix is detrimental in terms of financial expenses spent on computational resources and energy consumption, which should be reduced for ecological reasons. Therefore, there is a need to reuse these T-matrices once they have been calculated, and a fundamental request from the community concerns a standard data format that contains the T-matrix and unambiguous information about the corresponding object in terms of metadata. To respond to this demand, we describe here our efforts in the frame of DAPHONA project funded by BMBF to establish a data format and how to capitalize on it, using an infrastructure to archive and share T-matrices. Following the FAIR principles, we perceive a standard in HDF5 format, dedicate a database on a large-scale data facility, and provide search functionality available via the dedicated web server. Besides saving monetary and economic resources, this structure allows for a data-driven approach in this research field. It constitutes the first step in solving forward and inverse design problems based on the correspondence between the T-matrix and the geometry of the object with the help of machine learning.
Vapor deposition encompasses a vast array of techniques ranging from chemical vapor deposition (CVD) processes like metal-organic vapor phase epitaxy (MOVPE) to physical vapor deposition (PVD) processes like pulsed laser deposition (PLD). These processes are used within a diverse set of industries to deposit thin films and coatings for everything from television screens to corrosion protection. In order to further develop and apply these techniques it is crucial that we share a common understanding of the various terms and concepts we use to record and describe these processes. Therefore, we present an ontology for vapor deposition which extends the work of the Chemical Methods Ontology (CHMO), which is based on the Ontology for Biomedical Investigations (OBI), which in turn is based on the Basic Formal Ontology (BFO). In addition to the ontology development, we show how it has been practically implemented in the NOMAD repository and electronic lab notebook (ELN) as part of the NFDI project FAIRmat. Specifically, we show the implementation of MOVPE, PLD, molecular beam epitaxy (MBE), (reactive) sputter deposition, and thermal deposition techniques as well as how this implementation facilitates the interoperability and reusability of process data between co-workers, labs, and institutes on an international level.
The physical sciences community is increasingly taking advantage of the possibilities offered by modern data science to solve problems in experimental chemistry and potentially to change the way we design, conduct, and understand results from experiments. Successfully exploiting these opportunities involves considerable challenges. Here, we will present a perspective on the importance of data science and automated approaches in energy materials. We will focus on experimental co-design and its importance to experimental chemistry. We provide examples of how data science is changing the way we conduct experiments, and we outline opportunities for further integration of data science and experimental chemistry to advance these fields. Specific case studies will be related to the generation of solar fuels devices via artificial photosynthesis.
Introduction
The acquisition and storage of experimental data in the field of catalysis according to the FAIR principles (Findable, Accessible, Interoperable, and Reusable) necessitates the automation and digitization of experimental setups. In this work, we present our local solutions, in which we have integrated the concept of Standard Operating Procedures (SOPs) into automation workflows to enable the reproducibility and comparability of experimental data. The data are stored in a database, which accepts various data types and is easily accessible via its API (Application Programming Interface). The linking of entries, which is displayed in knowledge graphs, makes it possible to find and reuse the data and to track the history of projects.
Results
Automated systems have been developed to cover different cases, including automatically performed experiments and manually performed experiments. These systems consist of the following components:
(i) EPICS as a control system software, (ii) database (FHI Archive), (iii) EPICS Archiver Appliance for storing time series data, (iv) Phoebus software for creating graphical user interfaces (GUIs), (v) Python/Bluesky/Jupyter notebooks for creating automation and analysis scripts, and finally (vi) an S3 storage for long-term backup of experiment data.
An example of a fully automated setup following our concept is a test reactor for ammonia decomposition, which is able to perform experimental steps automatically according to a method that can be entered via a special GUI and stored in the database. In the case of commercial devices that cannot be automated, we present solutions for automatic acquisition of the output files, and uploading to the database, e.g. for data from electron microscopy (Talos, ThermoFisher) and gas chromatography (Agilent). Metadata and method data for manual experiments can be saved in JSON format by entering them into special interactive GUIs.
The advancement of digitalization in catalysis and other scientific domains is marked by a transition from paper-based documentation to electronic lab notebooks, standardized protocols, and experiment automation. This shift promises enhanced reproducibility, comparability, and overall scientific progress. However, at the moment the field of catalysis still lacks universal standards for documenting results and centralized repositories for storing, sharing, and accessing research data. In response, we are adapting an existing open-source software, NOMAD, originally designed for computational data, to accommodate experimental and heterogeneous catalysis data. Through NOMAD, tailored tools for data publication and search are provided to catalysis researchers.
Data organization in NOMAD involves the creation of entries for each catalyst sample and activity performed on that sample. References between these entries and overview workflow views facilitate retrospective research tracking. A mixture of predefined templates for heterogeneous catalysis datasets and customizable entry schemas are available to upload catalysis research data. Functions to directly import and visualize measurement data from different file formats such as csv, excel and hdf5 exist in these schemas. Semantic enrichment by annotating quantities with links to concepts defined in ontologies or vocabulary such as Voc4Cat facilitates advanced and interoperable research data management including AI application in catalysis related research.
Additionally, NOMAD features the Heterogeneous Catalysis Explore App, allowing for the aggregation and visualization of all catalysis related data. In this app, a set of customizable widgets summarize both text-based and numerical data, with a focus on reaction and catalyst properties, aiding in visual analysis and filtering.
With these developments in NOMAD we address the need for a comprehensive repository for experimental catalysis data and the standardization of machine-readable, structured data publication in the field. Our efforts should facilitate the publication of FAIR and open catalysis data and streamline literature searches and AI-assisted data analysis in the field.
A transition from polluting fossil fuels to cleaner energy sources is underway. However, the intermittent nature of renewables such as solar and wind, dependent on fluctuating environmental conditions, presents a challenge for maintaining a reliable energy supply. Water electrolysis offers a solution by employing excess renewable energy to split water into $\mathrm{H}_2$ and $\mathrm{O}_2$, which can then be converted back to electricity on demand via fuel cells.
Water electrolysis occurs via two half-reactions: the oxygen evolution reaction (OER) at the anode (Eq. 1) and the hydrogen evolution reaction (HER) at the cathode (Eq. 2).
\begin{alignat}{3}
&2\mathrm{H}2\mathrm{O}\rightarrow\mathrm{O}_2(g)+4\mathrm{H}^+ + 4\mathrm{e}^-&&\quad E^0=1.23\,\space\,\mathrm{V}\mathrm{RHE}\quad\text{(1)}\
&2\mathrm{H}^++2\mathrm{e}^-\rightarrow\mathrm{H}2(g)&&\quad E^0=0.00\,\space\,\mathrm{V}\mathrm{RHE}\quad\text{(2)}
\end{alignat}
The OER, limited by sluggish kinetics, currently relies on costly Ir$ \mathrm{O}_2$ catalysts which lack efficient atom economy, hindering wide-scale adoption.[1] Transition metal complexes (TMCs), with superior activity and improved atom economy, are promising but face stability issues.[2] Further studies are needed to design robust and active TMC catalysts for the OER.
In this presentation, we will introduce a database of candidate TMC complexes for the OER. This database is constructed using a bottom-up approach. Co, Cr, Fe, Mn, and Ru metals are combinatorically coordinated with bidentate and tridentate ligands forming unique entries within the database. Ligands were extracted from TMCs present in the Cambridge Structural Database.[3] These ligands were subsequently filtered to target instances suitable in the OER. We envisage this database enabling the discovery of robust TMCs with enhanced catalytic performance.
Computational databases are pivotal in modern chemistry, enabling the advanced data-driven exploration of chemical space. Transition metal complexes are a particularly versatile class of molecules due to their tunability of metal center and coordinating ligands, offering broad applications in therapeutics, catalysis and supramolecular chemistry. However, exploring the vast chemical space of possible molecular complexes remains challenging due to the need for efficient algorithms to generate realistic molecular complexes tailored to specific applications.
In this contribution, we address these challenges by introducing a new Python library named DART (“Directed Assembly of Random Transition metal complexes”)[1]. DART contains a dataset of 41,018 ligands extracted from 107,185 complexes recorded in the Cambridge Structural Database. Using these ligands, the algorithm assembles 3D structures of novel molecular complexes in a high-throughput fashion by combining multiple ligands with a specified metal center. In order to target specific chemical spaces, users can refine the input ligands by applying a selection of powerful ligand filters. All options in DART are set using straightforward yaml input files - making DART accessible to everyone independent of Python expertise and democratizing chemical modelling. As a minimal example, after downloading DART it is a matter of minutes to generate 1000 structures of neutral square-planar Pd(II) complexes with two randomly selected N-O donor ligands with charge of –1, which do not contain any methyl groups.
Overall, we expect our workflow to contribute to a rational approach to high-throughput screening and the generation of new databases of transition metal complexes shared FAIRly to support the accelerated discovery of new molecular complexes for targeted applications.
References:
[1] Sommer, T.; Clarke, C.; Kleuker, F.; García-Melchor, M. Manuscript in preparation.
The utilization of data intelligence tools presents numerous advantages for scientists and holds significant potential to streamline and expedite scientific endeavors across various domains. Specifically, research data infrastructures must address the opportunities and obstacles posed by data intelligence to ensure optimal support for their users and the broader scientific community. This talk will describe two general aspects within the realm of chemistry research data: (1) How does research data infrastructure benefit from the development and implementation of data intelligence tools and how can data intelligence support different areas of a research data infrastructure? (2) How can a research infrastructure contribute to promote the development of data analysis tools? What are suitable measures to design the future of chemistry work by promoting data intelligence in the long run? For both aspects, examples taken from the Chemotion ELN and the Chemotion repository will be utilized to describe current, already implemented tools and workflows, as well as those planned within NFDI4Chem. A highlight dealing with the half-automated curation of data will show the impact of data intelligence on efficient review options for scientific data. In this context, the impact of data intelligence on the establishment of automated synthesis platforms will be discussed.
Atom Probe Tomography (APT) is widely used for nanoscale structure and composition characterization across various disciplines, including materials science, geosciences, and biological sciences. Therefore, it is essential to have standardized workflows for analysis and post-processing that can combine software tools from different research communities in an interoperable manner. We demonstrate this by combining two distinct software tools, paraprobe-toolbox and CompositionSpace, developed within the FAIRmat and NFDI-MatWerk consortia, respectively, for APT data analysis. The integration of these tools enables the characterization of composition and structural features of interfaces in an alloy.
To ensure interoperability and reproducibility, these tools are made accessible through community code registries such as conda-forge. Furthermore, meticulous recording of details such as the used software and their corresponding versions, the computational environments, the execution order of different workflow steps, and provenance is required. These requirements lead to the need for FAIR computational workflows, which combine both FAIR software and data principles with extended requirements to ensure reproducibility, reuse, and repurposing. We employ pyiron as a workflow management tool and demonstrate its use for the analysis of data from APT experiments. pyiron provides a Jupyter notebook-based, user-friendly platform for users to compose workflows, significantly reducing the entry barrier for using and combining different software tools. Additionally, the workflows can be exported to other widespread workflow description languages such as Common Workflow Language, Snakemake, and NextFlow. The results from the APT analysis workflows are annotated according to an open description, and provenance records are maintained. Advanced visualization tools for APT results are also provided.
This initiative showcases the collaborative efforts of the NFDI-MatWerk and FAIRmat consortia, illustrating how user-friendly, interoperable, and reusable tools can be created. By establishing FAIR workflows for APT, we take a crucial step towards automating FAIR data production and advancing reproducible scientific analyses in this field.
Aiming at data-driven design of magnetic materials as a demonstration of using NOMAD to integrate automated workflows, metadata formulation, and machine learning, we elucidate how research data management can be implemented for first-principles calculations on magnetic materials. On the one hand, we have established workflows to perform high-throughput calculations on the intrinsic magnetic properties, including the magnetic ground state, saturation magnetization, Curie/Neel temperature, magneto-crystalline anisotropy, topological transport, and spectroscopic properties, for both crystalline and chemically disordered materials. On the other hand, extensive machine learning algorithms have been implemented to map out the structure-property relationships, and also to bridge to experimental simulations. We are going to show how the research data management can be performed for our data on NOMAD using pre-defined and custom schemas that it is Findable, Accessible, Interoperable, and Reusable (FAIR), with illustrative machine learning demos, facilitated by a local NOMAD Oasis at TU Darmstadt.
Structured data, in which properties of materials, systems, or devices, are tabulated in a systematic way is a foundation for the methodical optimization and design of novel materials or devices. One of the most widely known databases in materials science is the metal-halide perovskite solar cells database. While this database found widespread use it is difficult to update and extend as it has been manually curated.
Such manual curation requires tremendous labor and vigilant work and is thus not scalable.
Recent advances in large language models (LLMs) indicate that this manual work might at least partially be replaced using these models. A difficulty however is, that for scientific use cases we have high requirements on robustness. In addition, the relevant information is often dispersed across articles and partially in figures in tables, requiring more reasoning than just mere text extraction.
Given that there is already a large amount of extracted data in the perovskite database along with instructions for human extractors we can leverage this information to bootstrap an automatic and robust extraction pipeline based on large language models. The existing resources additionally, provide us with labeled information we leverage for systematic optimization.
Here we present an end-to-end pipeline that robustly extracts data, with more context than in the current database, in a scalable way. To develop an autonomous system, we couple our extraction pipeline to a paper crawler. With this, we can identify new relevant papers, extract relevant information, and then commit structured data with confidence scores into a staging area of the NOMAD [1, 2] database.
Our work provides a blueprint for the autonomous maintenance of datasets, which we believe is a key enabler for harnessing the collective knowledge of materials science that is currently in the dark.
Inorganic halide perovskites are promising for optoelectronic applications, offering greater thermal stability over hybrid counterparts but are prone to phase instabilities. Phase stability can be improved by compositional engineering, e.g., varying the Cs/Pb and I/Br ratio. Combinatorial vacuum coevaporation allows the investigation of the large compositional space of Cs(Sn,Pb)(I,Br)3 in the search for lead-free absorber layers of high electronic quality and stability.
To accelerate material exploration, we have automated the characterization and are able to characterize 600 samples on a 5 cm x 5 cm sample library by UV-Vis, PL, TRPL, conductivity, and XRF within one day after sample deposition in a glove-box based custom-built evaporation system. All measurements are performed either in an inert atmosphere or vacuum, preventing ambient-induced degradation.
Data from our experiments are directly fed into a specially customized NOMAD Oasis platform, shaped to meet our research specifications. This data management system not only enables large-scale data integration but also produces data that is ready for ML/AI analysis . By generating ML/AI-ready datasets, we facilitate the evaluation of critical material properties such as absorption coefficient, band gap, photoluminescence quantum yield, quasi-Fermi level splitting, recombination lifetime, doping density, and carrier mobility. Coupled with device simulation and empirical models, this approach allows us to rapidly predict solar cell performance from these material properties, typically within a day of synthesis.
Our methodology emphasizes the interoperability of the data artifacts generated, by ensuring compliance with data models established by a broad community of specialists. Specifically, the data fed into NOMAD Oasis adheres to models and base classes developed as part of an ontology for vacuum deposition. This approach not only aligns with the FAIR-data principles but also facilitates the reuse of data across various platforms and applications.
New materials are conventionally developed via trial and error in laboratory experiments.
This process is in general slow and involves significant resources and research eEorts.
Furthermore, it can overlook potential candidates, properties, or business-case criteria
related to their use. Computational simulation methods can help solve these problems by
accelerating the screening process and reducing costs. However, applying these methods
requires expert knowledge – uncommon in most industries – not to mention that they do not account for business criteria of utmost importance for industrial development.
We address this need by developing a materials informatics platform that allows our
industrial partners to screen out and find new materials for their applications with little to no prior knowledge of theoretical methods. Our solution combines atomistic simulations –
from density-functional theory to molecular dynamics – and machine learning models with
cost and sustainability data to oEer a holistic solution to materials screening. Our scoring
and ranking algorithms compare, and rate materials data based on chemical, physical, as
well as sustainability, and cost-related criteria. The technology behind our success has been developed at the German Aerospace Center (DLR), which ExoMatter spun out of.
We highlight the path from first-principles data to usable, application-related materials
properties, relevant for industrial applications. We show exemplary use cases in the areas
of carbon capture, polymer development and ceramics, and highlight our first steps towards an open materials data platform for science and industry.
While rapid exploration and optimisation of solution-processable materials in self-driving laboratories (SDLs) is advanced, adapting these approaches for inorganic materials using physical vapour deposition (PVD) presents challenges due to increased experimental complexity and higher time and energy demands for sample production. It is thus critical that the SDL’s underlying algorithms learn as much as possible about the parameter space of the new materials from as few experiments as possible.
We are developing an SDL for exploring new inorganic optoelectronic materials, with two partially conflicting aims: to produce better knowledge of the new materials, and to speed up the optimisation of their properties. Our proposed end-to-end automated workflow uses magnetron co-sputtering to deposit combinatorial thin films, a thermal post-process to convert them, and subsequent analysis by various techniques. We believe this relatively fast, flexible PVD process will facilitate rapid materials exploration that addresses the speed/knowledge generation trade-off.
Here, we focus on the use of machine learning to characterise co-sputtering processes as a function of process settings, to automatically define settings for achieving specified targets, such as deposit composition. Process data including deposition-rate feedback is used to first eliminate totally unsatisfactory process conditions (via active learning) and then build a model of the remaining four-dimensional process parameter space. Here we are using Gaussian process regression and Bayesian optimisation with active learning to update the model by querying the most informative points to be tested in the experiment. In this contribution we compare different acquisition functions including negative integrated posterior variance and Bayesian active learning by disagreement, to learn the space in as few queries as possible. A geometrical model of the sputtering flux is incorporated, to predict film compositions. We also present other aspects of the workflow including our experiences using the NOMAD database for materials-processing data.
State-of-the-art Bayesian optimization algorithms have the shortcoming of relying on a rather fixed experimental workflow. The possibility of making on-the-fly decisions about changes in the planned sequence of experiments is usually excluded and the models often do not take advantage of known structure in the problem or of information given by intermediate proxy measurements [1-3]. We hypothesize that an extended Bayesian optimization procedure, with surrogate models and acquisition functions that can flexibly choose to modify the workflow on the fly, will improve the performance of state-of-the-art methods for optimization in self-driving labs.
To address these limitations, we developed a surrogate model composed of a sequence of Gaussian processes, that can take advantage of the modular structure of experimental processes to handle sparse datasets where only partial information (proxy measurements) is available [4]. We implemented an acquisition function, based on a mixture of expectation improvement and upper confidence bound, that allows the optimizer to selectively sample from individual sub-processes. Finally, we devised a synthetic dataset generator to simulate multi-step processes with tunable function complexity at each step, to evaluate the efficiency of our model compared to standard BO under various scenarios.
We conducted experiments to evaluate our model across nine distinct scenarios. In all scenarios our multi-step optimizer outperformed the benchmark methods, demonstrating superior performance in terms of both the quality of the optimum and in terms of convergence speed. This advantage is particularly evident in scenarios where the complexity of the first step exceeds that of the second step. We are currently in the process of validating our results on real-world datasets.
[1] Wu et al. 2023, JACS 145 (30).
[2] Seifermann, et al. 2023. Small Methods, 7(9).
[3] Jenewein et al. 2023. Journal of Materials Chemistry A, 12(5).
[4] Torresi et al. 2024, submitted.
Infrared Spectroscopy (IR) is crucial in heterogeneous catalysis for identifying active sites, yet existing simulations lack comprehensice peak broadening output. We propose an application to generate complete spectra from Density Functional Theory (DFT) data, facilitating comparison with experimental results. Built on CaRMeN, it manages data in an SQL database, ensuring efficiency and security. The frontend employs React and Relay for a user-friendly interface. Our tool integrates experimental and DFT-simulated IR spectra, allowing for metadata inclusion and responsive search. Users can organize spectra for comparison and adjust them, with options for transmission and absorbance data. Pre-defined Views aid in common adsorbate-catalyst analysis.
Most current explainable AI methods are post-hoc methods that analyze trained models and only generate importance annotations, which often leads to an accuracy-explainability tradeoff and limits interpretability. Here, we propose a self-explaining multi-explanation graph attention network (MEGAN) [1]. Unlike existing graph explainability methods, our network can produce node and edge attributional explanations along multiple channels, the number of which is independent of task specifications. This proves crucial to improve the interpretability of graph regression predictions, as explanations are intrinsically connected to their effect on the predictions. This makes MEGAN a successful attempt to escape the accuracy-explainability dilemma of post-hoc explanation models.
We first validate our model on a synthetic graph regression dataset with known ground-truth explanations. Our network outperforms existing baseline explainability methods such as GNNExplainer [2] for the single- as well as the multi-explanation case, achieving near-perfect explanation accuracy during explanation supervision. We demonstrate our model’s capabilities on multiple real-world datasets, e.g. molecular solubility prediction. We find that our model produces sparse high-fidelity explanations consistent with human intuition about those tasks.
Finally, we demonstrate in a real-world task in the area of drug discovery that the MEGAN model can be combined with counterfactual approaches for predictive in-silico molecular design, which we validated experimentally [3]. To our knowledge, this is the first example of rational molecular design with explainable machine learning models.
[1] J. Teufel, L. Torresi, P. Reiser, P. Friederich, MEGAN: Multi-explanation Graph Attention Network, xAI Conference 2023, 338–360.
[2] Z. Ying, D. Bourgeois, J. You, M. Zitnik, J. Leskovec, GNNExplainer: Generating Explanations for Graph Neural Networks, NeurIPS 2019.
[3] H. Sturm, J. Teufel, K.A. Isfeld, P. Friederich, R.L. Davis, Mitigating Molecular Aggregation in Drug Discovery with Predictive Insights from Explainable AI, arXiv preprint arXiv:2306.02206 2024.
NOMAD [nomad-lab.eu] [1, 2] is an open-source data infrastructure for materials science data. NOMAD already supports an array of computational codes and techniques, with over 60 parsers that automatically extract essential (meta)data from the raw output of standard calculations. Traditionally, the NOMAD repository has focused on contributions from DFT calculations, accumulating over 12.5 million such entries. More recently, this framework has been expanded considerably, now supporting classical molecular dynamics simulations, as well as complex simulation workflows. In this context, a variety of new features have been implemented into NOMAD, including a schema for defining molecular topologies and system hierarchies. In this contribution, we will introduce NOMAD in the context of soft matter simulations, demonstrating some basic functionalities and its potential for improving the data management standards within the classical simulation community through its adherence to the FAIR principles (Findability, Accessibility, Interoperability, Reusability) [3].
[1] Scheidgen, M. et al., JOSS 8, 5388 (2023).
[2] Scheffler, M. et al., Nature 604, 635-642 (2022).
[3] Wilkinson, M. D. et al., Sci. Data 3, 160018 (2016).
Advancements in materials science are significantly dependent on the detailed characterization of samples, which in turn generates complex measurement data. This poses challenges in data management, notably in metadata preservation and the need for extensive manual processing, often exceeding the expertise of researchers. The FAIR principles offer a pathway towards resolving these issues through standardization and well-documented metadata, yet the adoption across materials science has been slow due to the lack of a unified community approach.
The NOMAD platform addresses this gap by extending its repository capabilities to include experimental data, facilitated by the NOMAD measurement plugin. This tool enhances data interoperability and reusability, simplifying the management of experimental measurement data, exemplified here with X-ray Diffraction (XRD) data. It enables automatic data ingestion, standardization, and accessibility in open formats, aligning with the FAIR principles and promoting a collaborative ecosystem in materials science.
The plugin’s foundation in NOMAD’s base section data model guarantees cross-entry interoperability, allowing for the development of specialized tools. By leveraging Python, it supports extensive automation in data processing and visualization, fostering community-driven development through customizable data schemas.
The NOMAD measurement plugin not only exemplifies a user-friendly approach to FAIR-compliant data management but also encourages collaborative innovation within the materials science community. Its integration with NOMAD’s analysis tools further ensures the data’s readiness for advanced applications, marking a significant step towards standardized, collaborative research data management.
We introduce NOMAD CAMELS [1] (Configurable Application for Measurements, Experiments, and Laboratory Systems), an innovative open-source measurement software designed to capture FAIR data that is fully self-describing in NeXus format. This enables native integration of CAMELS’ data into research data management tools such as NOMAD or eLabFTW. CAMELS empowers users to define measurement protocols through an intuitive graphical interface, eliminating the need for programming skills or in-depth knowledge of instrument communication. Originally developed for solid-state physics, CAMELS offers great flexibility in controlling a diverse range of measurement instruments within dynamically changing experimental setups. The user-defined protocols are translated into standalone executable Python code, ensuring complete transparency in the execution of measurement sequences. Python's utilization enables the usage of numerous sophisticated libraries and its ongoing improvement and worldwide community provide a framework allowing CAMELS to grow with new developments.
[1] https://joss.theoj.org/papers/10.21105/joss.06371
The rise of digitalization has significantly reshaped scientific practices, positioning research data as a valuable asset. New research paradigms have emerged that extend the use of these data beyond their original research purposes. As a result, proper data preservation in line with the FAIR principles,[1] as well as the legal aspects relevant to the preservation and reuse of these data, have grown in importance.
In this contribution, we explore various legal aspects of research data management (RDM) that are relevant to each stage of the data lifecycle.[2] We address data-related legal considerations commonly found in contracts and discuss a range of intellectual property rights that are relevant to research activities. This discussion includes copyright, relevant licenses, database rights, and the exemptions that allow the use of copyrighted works in scientific research. In addition, legal considerations related to open access, international data transfer, and cybersecurity are reviewed to provide a comprehensive overview of the legal landscape in RDM.
Reference:
[1] M.D. Wilkinson et al., "The FAIR Guiding Principles for scientific data management and stewardship," Sci. Data 3, 160018 (2016).
[2] S. Nakhaie, A. E. Mansour, K. Helbig, M. Bierwirth, M. Aeschlimann, and C. Draxl, “FAIRmat Guide to Legal Aspects in Research Data Management”, FAIRmat (2024) https://zenodo.org/records/11083303.
A key challenge in experimental high-resolution microscopy is the real-time interpretation of the observed images in conjunction with the parameters adjusted by the experimenter during data acquisition, e.g. to obtain a certain contrast. The parameter space of candidate structures, experimental parameters, and resulting image contrast can be vast and complex, often requiring a scientist who is well-trained in performing image simulations and a lot of trial and error to come up with a plausible interpretation of the observation. To make such expert simulations accessible to anyone, we introduce LLMicroscopilot, a chatbot tool powered by a large language model, that searches materials databases for suitable structures and simulates various types of microscopy images from them, without requiring the user to know how to operate either the database search, or the simulation software. It recommends suitable simulation parameters for obtaining high-quality results, recovers from errors in code execution, and refines results in dialog with the user. We demonstrate the capability of Microscopilot to operate transmission electron microscopy (TEM) and scanning tunneling microscopy (STM) simulation software back-ends, and more. Additionally, we highlight the potential for mining the memory of microscopy parameters that this tool is capable of building up, e.g. by recommending parameters that have led to accepted results in previous uses.
Electronic Laboratory Notebooks (ELNs) are crucial for moving research data from paper to digital formats, streamlining lab workflows and digitizing data. This study examines integrating ELNs into Research Data Management (RDM) platforms like NOMAD, focusing on challenges like user acceptance and data structuring.
ELNs need to be user-friendly and structure data effectively for integration into RDM platforms, where structured data is vital for interoperability and advanced analyses. NOMAD aims to harmonize data across sources, requiring structured data from ELNs, a challenge due to their typically unstructured nature.
Our research presents how NOMAD enhances ELN integration, facilitating structured data creation and improving user engagement. NOMAD’s data processing capabilities and plugin mechanism enable the conversion of raw files into structured entries, promoting automation and acceptance.
Moreover, NOMAD allows for the creation of highly structured ELNs from predefined schemas and integrates third-party ELNs through templates for structured data entry. This approach balances user preferences with the advantages of RDM tools, advancing structured data collection and adhering to FAIR data principles.
This study highlights the role of ELNs integrated with platforms like NOMAD in modernizing research data management and enhancing data usability.
At the Helmholtz Association, we strive to establish a well-formed harmonized data space, connecting information across distributed data infrastructures. This requires standardizing the description of data sets with suitable metadata to achieve interoperability and machine actionability.
One way to make connections between datasets and to avoid redundancy in metadata is the consistent use of Persistent Identifiers (PIDs). A lot of information within the metadata such as people, organizations, projects, laboratories, repositories, publications, vocabularies, samples, instruments, licenses, and methods should be commonly referenced by PIDs, but not for all of these agreed identifiers exist yet.
Typically, researchers who are publishing datasets are also tasked with compiling the metadata for those datasets. However, researchers are usually not in charge of a lot of information that should be part of the documentation of a dataset. They often have to rely on information they receive from other sources, e.g. technicians, responsible for the measuring devices or librarians, who are experts in assigning licenses. Starting from PID Systems ROR, ORCID, IGSN, PIDInst, DataCiteDOI and CrossRef DOI we suggest to share the load, and assign certain expert stakeholder groups responsibility to maintain specific information and to conduct certain tasks within the research data management (RDM) workflow.
The conclusions from this process do not only affect the implementation of PID metadata, but may also be used for the harmonization of vocabularies, digital objects, interfaces, licenses, quality flags and others, in order to connect our global data systems, to redefine stakeholder responsibility and to ultimately reach the data space.
Recently, funding agencies have begun to require sections on research data management in grant applications and the submission of a detailed Data Management Plan (DMP) during the initial phase of a funded research project. These DMPs are set as milestones to be achieved for a successful research project. Scientists often view DMPs as a burden and additional work that distracts them from active research. This is due to different requirements from different funders, lack of established routines for writing DMPs, and lack of tools and services for efficient research data management.
We analyzed the DMP guidelines provided by the European Union for its Horizon Europe and ERC (European Research Council) grants, as well as those of the German Research Foundation (DFG). We summarized the requirements and topics to be covered, contrasting similarities and differences while keeping an eye on the different details.
This poster will provide an overview of the basics of writing a DMP and will emphasize that writing is only a small part. The main work is preparation, thorough discussion, and decision making about how to manage the data. The groundwork includes documentation and data quality, storage and archiving, legal aspects, data sharing, and accessibility, to name a few. Some funders require researchers to apply the FAIR (Findable, Accessible, Interoperable, and Re-usable) data principles for research data management. We will present our hands-on workshop for planning and writing DMPs and provide examples of how NOMAD, a web-based open source service, can support research data management according to the FAIR principles. Thorough data management planning in the early stages of a research project can prevent many of the hurdles that can arise in relation to the above issues. Therefore, a well-thought-out data management routine is critical to the success of the project.
The vast chemical landscape of metal–organic frameworks (MOFs) offers a rich array of compositions, structures, and potential applications.[1] Advancements in Artificial Intelligence (AI) and computer-assisted techniques have not only enhanced MOF discovery but also the field of MOF synthesis.[2] In this presentation, I will offer an experimentalist’s perspective on integrating AI into MOF synthesis and discovery.
In selected examples, I will present closed-loop AI strategies to determine optimal growth conditions for Surface-anchored MOFs (SURMOFs),[3] resulting in enhanced crystallinity, uniform orientation, and low surface roughness.[4] Furthermore, I will discuss how automated data extraction, in combination with machine learning, can be employed to accelerate the synthesis of MOFs.[5] Moreover, the talk will highlight the role of research data management tools in enhancing these data-driven approaches.[6]
References
[1] (a) H. Furukawa et al. Science 2013, 341, 1230444; (b) S. Kitagawa et al. Angew. Chem. Int. Ed. 2004, 43, 2334.
[2] (a) H. Lyu et al. Chem, 2020, 6, 2219; (b) S. M. Moosavi et al. Nat Commun. 2019, 10, 539; (c) P. Z. Moghadam et al. Nat Energy 2024, https://doi.org/10.1038/s41560-023-01417-2; (d) H. Daglar et al. ACS Applied Materials & Interfaces 2022 14, 32134; (e) M. Jalali et al. Nanomaterials 2022, 12, 704; (f) Z. Zheng et al. Angew. Chem. Int. Ed. 2023, 62, e202311983; (g) Y. Luo et al. Adv. Mater. 2019, 31, 1901744.
[3] O. Shekhah et al. Chem. Soc. Rev., 2011, 40, 1081.
[4] (a) L. Pilz et al. Adv. Mater. Interfaces 2023, 10, 2201771; (b) L. Pilz et al. J. Mater. Chem. A, 2023, 11, 24724.
[5] L. Glasby et al. Chemistry of Materials 2023, 35, 4510; (b) P. Kalhor et al. Adv. Funct. Mater. 2024, 2302630; (c) Y. Luo et al. Angew. Chem. Int. Ed. 2022, 61, e202200242;
[6] C-L Lin et al. ChemRxiv. 2023 https://doi.org/10.26434/chemrxiv-2023-2dd4c
Integrating artificial intelligence (AI) with metal-organic frameworks (MOFs) and highly versatile and structurally diverse materials heralds a new era in material science, offering groundbreaking solutions to longstanding challenges in engineering and data analytics. MOFs, known for their exceptional porosity and customizable frameworks, have shown promising applications across various fields, including gas storage, separation processes, catalysis, and drug delivery. However, accurately predicting and analyzing the accessibility of guest molecules within these frameworks is a critical determinant of their functional performance.
Our research introduces MOFGalaxyNet1, a novel AI framework designed to significantly enhance the prediction and analysis of guest molecule accessibility within MOFs, moving beyond the limitations of traditional methodologies. By employing a unique combination of social network analysis (SNA) and graph convolutional networks (GCNs), MOFGalaxyNet offers a new perspective on the structural analysis of MOFs. This approach treats MOFs as dynamic networks of nodes and edges, where nodes represent the organic linkers or metal ions, and edges depict the connections between them. This paradigm allows for an in-depth understanding of the structural features governing guest molecule accessibility. It facilitates the transformation of MOFs' structural data into a numerical vector representation encapsulating their connectivity and topology.
One cornerstone innovation of MOFGalaxyNet is the application of GCNs to predict the pore-limiting diameter (PLD) of MOFs, a critical parameter for determining guest accessibility. This capability significantly advances existing machine learning models, offering a faster, more efficient means of screening MOFs for specific applications. Through a comprehensive dataset of various MOFs characterized by their unique structural properties, MOFGalaxyNet has demonstrated superior performance in predicting PLDs with remarkable accuracy.
The implications of our findings extend beyond the immediate advancements in MOF research. By highlighting the potential of AI to revolutionize material discovery and characterization, MOFGalaxyNet exemplifies the broader significance of integrating AI with material science.
Hydride materials that can reversibly abs-/desorb hydrogen have been intensively investigated due to their potential as a hydrogen storage medium and functional materials for many applications. The dynamic reaction between hydride/hydride-forming material and the gaseous phase is complex and has several intermediary processes, making the insightful description of these phenomena impractical, especially for calculations. However, such hydride material formation/decomposition is assumed to occur in steps, and the slowest step characterizes the overall dynamic process. This step is called the rate-limiting step (RLS). With the help of experimental data and empirical equations that describe each different RLS, it is possible to model the most relevant processes better. In addition, differential equations can be obtained for the numerical design and optimization of the storage systems. However, this previous approach requires a time-consuming analysis of the experiments. Assessment is only possible with expert knowledge of materials behavior to determine the most likely RLS of each material at a specific combination of temperature and pressure. In order to overcome these fundamental limitations, a machine learning approach is used here for the first time, in which surrogate models are learned to describe the materials behavior.
This work conducts kinetic measurements with an AB2 hydride-forming alloy (Hydralloy C5) in different temperature and pressure conditions. The gathered data is employed to develop non-supervised machine learning models to identify the RLS. The results show an accuracy of more than 80% considering only the best-ranking model and close to 97% when very close second and third matches based on the R² values of the original analysis are considered. The application of machine learning methods to the kinetics of hydrides facilitates and accelerates the development of a streamlined, highly automated determination of the kinetic parameters and the kinetic equations for application in hydride-based system designs.
Modern chemical technology makes extensive use of formulated products, from flavours and fragrances to surfactants and resins. These products are traditionally created and optimised using trial and error, an inefficient and costly process. This situation arises in part due to the complexity of the task. Understanding and accounting for the huge number of (inter)molecular interactions in a design task is currently a huge challenge. The Big Chemistry Consortium aims to transform formulation from an art to a science-based technology by establishing the RobotLab, an autonomous, self-driving laboratory combining AI, chemical data and high-throughput experimentation. Our consortium is spread across several institutions in The Netherlands (Radboud University Nijmegen, TU Eindhoven, AMOLF, Rijksuniversiteit Groningen and Fontys Hogeschool), each contributing to a pool of experimental methods, data and expertise. In this talk, I will present a selection of our initial investigations in high-throughput data collection, how we are approaching AI methods such as chemical language models for property prediction, and how we are working towards establishing efficient and secure data sharing methods within the consortium.
ALBA synchrotron has pledged to follow the FAIR data management principles by which results produced by academic users will be available to the public. One key step of this commitment is to standardize the process by which data are stored. At ALBA this process is being made by rigorously following NeXusFormat application definitions that determine which metadata are essential to replicate the experimental results. Hence, in this talk we will discuss the status of our NeXusFormat implementation and show the benefits of adopting it, including the features available for the users when accessing their data through the ALBA's ICAT catalogue.
Surprisingly, despite the rapid progress of machine learning in materials science, the prediction of optical spectra for crystalline materials remains underexplored, although this gap presents an opportunity to discover novel or tailored materials for various optical applications, including photovoltaic systems, photocatalytic water splitting, epsilon-near-zero materials, optical sensors, and light-emitting devices.
The review "Roadmap on Machine Learning in Electronic Structure" [1] highlights this absence, as optical spectra - such as the dielectric function $\varepsilon(\omega)$ or the refractive index $n(\omega)$ - receive little attention. The section on deep learning spectroscopy deals mainly with vibrational spectra, which are easier to calculate due to their dependence on nuclear motions.
In order to investigate the potential of predicting frequency-dependent optical spectra of crystalline solids with state-of-the-art deep learning models, we have created a database using ab initio calculations. Our database contains the frequency-dependent dielectric tensors for 9,915 crystalline semiconductors and insulators, from which all other linear optical properties can be derived.
Using this database, we show that deep learning models commonly used in materials science can effectively learn the spectral properties of semiconductors and insulators. This achievement does not require significant changes to the model architecture or a large database, even when predicting a high-dimensional vectorial quantity instead of a scalar.
Our model achieves very good quantitative agreement when evaluated on the test set. The predicted spectra faithfully reproduce significant features without non-physical artifacts such as discontinuities, significant noise, or extreme values, despite the lack of forced continuity or smoothness.
[1] Kulik, H. J. et al. Roadmap on machine learning in electronic structure. Electron. Struct. 4, 023004 (2022).
In an era of rapid technological advancement and data proliferation, the ability to efficiently access and utilise scientific knowledge has become paramount. Here, we present the development of an advanced research assistant chatbot specifically designed to navigate and interpret scientific publications. Our approach uses the Retrieval-Augmented Generation (RAG) architecture, a state-of-the-art framework that combines the strengths of retrieval-based and generative machine learning models to improve information retrieval and response accuracy. The assistant chatbot is designed to assist researchers by providing concise summaries, extracting key information, and answering complex queries related to scientific documents. In addition, we will showcase its integration with Kadi4Mat, an open-source platform designed to efficiently manage research data and provide seamless access to a vast repository of research data. By linking our chatbot to Kadi4Mat, we ensure that users have access to up-to-date, high-quality research data and their own uploaded document set, thus providing a robust tool for scientific inquiry and, consequently, enabling more informed decision-making and accelerating the research process. We will demonstrate the effectiveness of our research assistant through a series of use cases, highlighting its ability to improve research efficiency and collaboration. This presentation aims to demonstrate the potential of combining advanced language models with comprehensive data management platforms to transform the landscape of scientific research support.
A lot of materials knowledge is obtained in an indirect manner, e.g. by fitting model parameters to data that is being acquired in some potentially very complex experiment. Electron microscopy data, for example, can be several 10s of GB; and especially for these very large sets of data, complex data analysis workflows (DAWs) must then be run, for extracting the materials property information that is being sought. In order to be used by researchers in the field, these DAWs are accompanied by an extensive user manual and often also a complex GUI, both of which must be updated every time a new feature is implemented in the DAW, or a new set of parameters controlling the DAW, has been discovered to improve performance. Before being able to utilize the functionality provided by the DAW, any person planning to apply it has to make himself / herself familiar with the specific sequence of commands, or the scripting commands, or the GUI operations that are required to perform the analysis, and it may not be very obvious to the person learning this operation, how to choose optimal parameters / settings. Together with the need to first install the software providing the DAW on a local computer, or server, the hurdle of having to first familiarize with its operation may prevent its use in many cases. Making the DAW available online with an LLM-based agent with access to the API documentation for resource-augmented generation (RAG) makes it usable for novice users immediately and reduces the effort to develop and maintain a GUI, keep the full documentation up to date, or train new users. Multiple and very different DAWs can be offered through the same interface. Example DAWs from the field of electron microscopy will be presented.